
Modernizing Pharmaceutical Quality Control Through UPLC, Automation, and Lifecycle Science
Mr. Hemant Patil, Pharmaceutical Professional
Affiliation: AscentCoAI Pharma
Correspondence: hemant@ascentcoaipharma.com
Keywords: UPLC, HPLC modernization, method consolidation, ICH Q2(R2), ICH Q12, data integrity, potentiometric titration, method development, method validation, Quality, lifecycle management, impurity profiling, mass balance, green chemistry.
Abstract
The pharmaceutical analytical landscape is undergoing a paradigm shift from compartmentalized, time-intensive methods toward integrated, high-efficiency analytical control strategies. Legacy High-Performance Liquid Chromatography (HPLC) methods, characterized by prolonged run times (30-90 minutes), are a significant source of operational vulnerability. These methods induce system stress, provoke deviations, and impede batch release, while their separation from assay, related substances (RS), and identification procedures triples validation efforts. This article presents a scientifically rigorous, regulatorily-aligned framework for modernizing pharmaceutical quality control. We advocate for the strategic consolidation of analytical procedures into single, robust Ultra-Performance Liquid Chromatography (UPLC/UHPLC) methods, supported by LC-MS for identification and the migration of classical titrimetry to automated potentiometry. Grounded in ICH Q2(R2), Q3A/B, and Q12 guidelines, this approach transforms quality control from a cost center into a strategic asset, delivering unparalleled gains in throughput, data integrity, cost efficiency, and scientific control throughout the product lifecycle, while significantly reducing environmental impact through minimized solvent consumption.
1. Introduction: The Hidden Costs and Scientific Limitations of Legacy Analytical Systems
For decades, pharmaceutical quality control has relied on a segregated model: one lengthy HPLC method for assay (potency), another often longer for related substance (RS) profiling, and potentially separate techniques for identification. Developed under historical technological constraints (limited pressure tolerances, older column chemistries), these methods remain validated but are misaligned with modern capabilities and expectations.
The extended run times of these methods are not benign. Continuous mobile phase flow over hours creates sustained backpressure, accelerating column degradation and causing seal fatigue, leaks, and pumpfailures.Retained residues can slowly crystallize within column frits, leading to unpredictable pressure spikes and the "ghost peak" phenomenon where inadequately eluted impurities appear in subsequent injections, compromising data integrity and batch validity. Each failure triggers a cascade of Out-of-Specification (OOS) investigations, deviation reports, and corrective actions, consuming hundreds of analyst and QA hours annually. Ironically, these failures are often misattributed to analyst error, while the root cause lies in an outdated method design.
From a scientific R&D perspective, these legacy methods often lack the sensitivity and resolution required for modern impurity control per ICH Q3A/B, risking incomplete mass balance and an inadequate safety profile. This article delineates a strategic blueprint for modernization, demonstrating that through method consolidation, advanced technology adoption, and rigorous regulatory science, laboratories can achieve a transformative leap in efficiency, reliability, and compliance
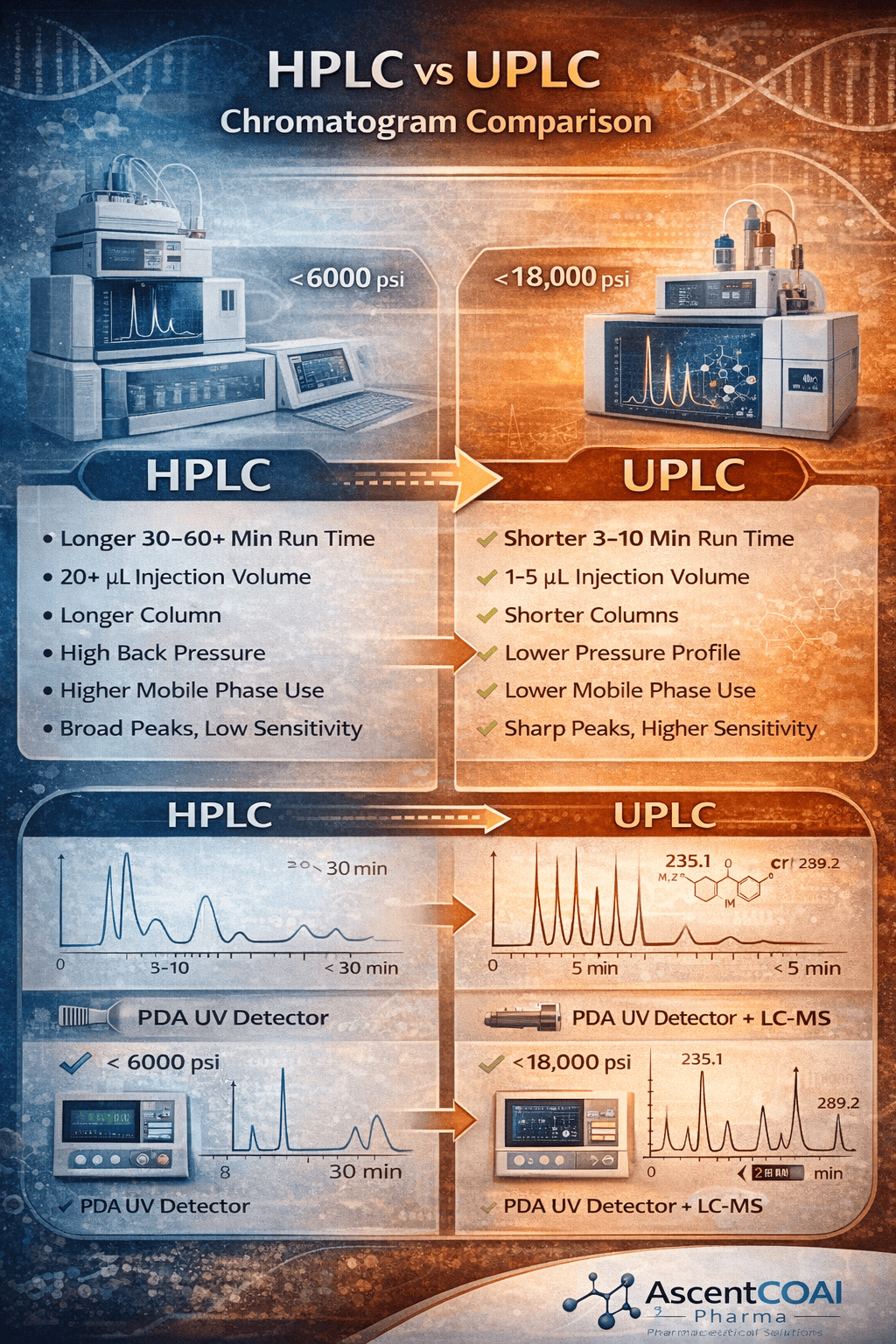 Figure 1: HPLC vs UPLC Chromatogram Comparison
Figure 1: HPLC vs UPLC Chromatogram Comparison
2. The Scientific Imperative for Method Consolidation and Technological Upgrade
2.1. Chromatographic Modernization: The UPLC/UHPLC Revolution
The core of the modernization strategy is the transition from traditional HPLC (≤ 6000 psi) to UPLC/UHPLC systems (15,000-21,000 psi) employing sub-2µm or advanced core-shell particle columns.
Scientific Basis and Advantages:
- The theoretical foundation is explained by the Van Deemter equation (H = A + (B/u) + C·u), where H represents plate height and u is linear velocity. A = Eddy diffusion (column packing heterogeneity), B/u = Longitudinal diffusion, C·u = Resistance to mass transfer. Sub‑2 µm particles drastically reduce the C term (resistance to mass transfer), allowing for optimal performance at significantly higher linear velocities without sacrificing efficiency. This translates into direct, quantifiable benefits:
- Dramatic Runtime Reduction: Achieves a 3- to 10-fold decrease in analysis time (e.g., from 45 min to 5 min), directly increasing throughput.
- Enhanced Sensitivity and Resolution: Sharper, taller peaks yield a superior signal-to-noise (S/N) ratio, lowering limits of detection (LOD) and quantification (LOQ) for low-level impurities. This is critical for comprehensive impurity control as per ICH Q3A/B.
- Feasibility of Single, Stability-Indicating Methods: The superior peak capacity and resolution enable the intelligent design of one method to simultaneously quantify the assay, profile all specified and unspecified impurities, and when coupled with a photodiode array (PDA) or mass spectrometer provide identification. This "triple-duty" approach is scientifically sound where molecule properties allow.
- Environmental Safety Benefits: UPLC methods typically reduce solvent consumption by 70-90% compared to traditional HPLC. This significantly decreases laboratory exposure to organic solvents (like acetonitrile and methanol), reduces hazardous waste generation, lowers carbon footprint from solvent production and disposal, and aligns with Green Chemistry principles and corporate sustainability goals.
Performance Comparison: Traditional HPLC vs. Modern UPLC/UHPLC Platforms
Parameter | Traditional HPLC | Modern UPLC / UHPLC + LC-MS | Scientific & Operational Impact |
Typical Run Time | 20–60 minutes | 3–10 minutes | 5×–12× faster throughput; enables rapid batch release and high-volume testing. |
Theoretical Plates | ~10,000–15,000 | ~50,000–150,000+ | Superior resolving power for complex impurity mixtures; enables method consolidation. |
Peak Capacity | Low to moderate | High | More components separated in a single run; supports comprehensive impurity profiling. |
Signal-to-Noise (S/N) | Baseline | 2×–5× higher | Enhanced sensitivity for trace impurity detection (LOD/LOQ); aligns with ICH Q3A/B thresholds. |
Solvent Consumption per Run | High (5–20 mL) | Low (1–3 mL) | >70% reduction in solvent cost and waste; supports green laboratory initiatives. |
System Pressure | Moderate (1,000–4,000 psi) | High (8,000–15,000+ psi), but for shorter duration | Reduced prolonged stress on seals and components, leading to improved system reliability. |
2.2. The Critical Role of Advanced Detection: LC-MS and Dual-Wavelength Strategies
•LC-MS/MS for Unequivocal Identification: The integration of mass spectrometry transforms identification from a tentative, retention time-based exercise into a confirmatory science. A validated LC-MS method provides molecular weight and fragmentation evidence for impurities, degradants, and the API itself, strengthening stability-indicating claims and supporting investigations. For complex molecules, a hyphenated UV-MS system can run the consolidated assay/RS method, collecting quantitative and qualitative data in one injection.
•Dual-Wavelength Detection for Mass Balance: To ensure complete impurity capture and justify mass balance a key ICH requirement—PDA detectors are essential. Employing dual or multiple wavelengths within the same chromatographic method compensates for impurities with differing chromophores, ensuring none are under-reported due to poor UV response at a single wavelength.
 Figure 2: Classical Titration vs Potentiometric Titration
Figure 2: Classical Titration vs Potentiometric Titration
2.3. Modernizing Wet Chemistry: From Subjective Titration to Automated Potentiometry
Wet chemistry methods are not exempt from modernization. Manual titrimetry, while historically reliable, is fundamentally dependent on visual endpoint interpretation, introducing analyst-to-analyst variability and subjectivity.
•Scientific Superiority of Potentiometry: Automated potentiometric titration replaces the human eye with an objective electrochemical sensor (pH or mV electrode). The endpoint is determined mathematically from the inflection point of the titration curve (dV/dmL), eliminating bias.
•Impact on Data Integrity and Accuracy: This transition ensures the procedure is executed identically every time. All parameters (sample weight, titrant, endpoint criteria) are locked in the software, creating a complete, audit-trailed electronic record that fulfills ALCOA+ principles. Precision (RSD) often improves from ±1-2% (visual) to <0.5% (potentiometric).
3. Regulatory and Scientific Justification: A Risk-Based, Lifecycle ApproachA prevailing misconception is that changing an approved method is prohibitively difficult. Modern ICH guidelines provide a clear, science-based framework for post-approval analytical improvement.
3.1. Grounded in ICH Guidelines
This modernization strategy is not only scientifically sound but is actively endorsed by contemporary regulatory frameworks:
•ICH Q2(R2) – Validation of Analytical Procedures: Explicitly recognizes that modern analytical techniques (including UPLC/UHPLC) may offer superior performance characteristics. As long as the modernized method meets or exceeds the validation criteria for the intended use, it is acceptable indeed, preferable for lifecycle management.
•ICH Q3A/B – Impurities in Drug Substances and Products: Requires comprehensive impurity profiling with defined reporting, identification, and qualification thresholds. A consolidated UPLC-MS method directly enables robust impurity profiling with enhanced sensitivity, ensuring compliance with these thresholds and supporting a complete mass balance. This reflects improved control, not method failure.
•ICH Q12 –Pharmaceutical Product Lifecycle Management: Emphasizes continuous improvement and the use of established conditions (product and process understanding) to support post-approval changes with reduced regulatory burden. Demonstrating equivalency or superiority of a modernized method, especially through comparative validation, aligns perfectly with Q12's philosophy. For commercial products with robust historical data, this can often be managed through an appropriate comparability protocol or change notification.
•ICH Q14 – Analytical Procedure Development: Promotes a science and risk-based development approach, inherently supporting robust, efficient, and future ready methods designed for the entire lifecycle.
 Figure 3: The Analytical Renaissance - Modernizing Pharmaceutical Quality Control
Figure 3: The Analytical Renaissance - Modernizing Pharmaceutical Quality Control
3.2. Demonstrating Equivalency: Comparative Validation and Correlation Studies
For commercial products with established legacy methods, the transition to a modernized method must be scientifically justified:
•Parallel Testing: Run a diverse, representative set of samples (including stability, stressed, and spiked samples) on both the legacy HPLC method and the new UPLC method. Demonstrate that the UPLC method achieves equivalent or better separation, reproducibility, accuracy, and limit of detection/quantification (LOD/LOQ).
•Statistical Equivalence: Employ statistical methods (e.g., two one-sided t-tests [TOST], Bland-Altman plots) to confirm that the mean results and variability from both methods fall within predetermined, scientifically justified acceptance criteria.
•Enhanced Impurity Detection: Demonstrate that the UPLC-MS method can detect and quantify all impurities previously observed, plus any new or co-eluting peaks that were missed by the legacy method, thus establishing superiority in impurity profiling.
This evidence package, combined with a full validation of the UPLC method per ICH Q2(R2) (specificity, linearity, accuracy, precision, range, robustness), provides a compelling technical justification for regulatory submission. For products with well-characterized impurity profiles and existing regulatory flexibility (e.g., via established design space or product lifecycle management frameworks), such changes may be implemented via post-approval change protocols, reducing the regulatory burden.
3.3. Addressing Complexity: When is Consolidation Appropriate?
Ideal Candidates: Molecules with well-separated UV-active peaks, straightforward matrix effects, and defined impurity profiles are excellent candidates for a consolidated method approach.
Precaution and the Role of R&D: This strategy is not universally "plug-and-play." For critical molecules with extremely complex profiles (e.g., chiral mixtures, labile compounds), initial equivalency may not be achieved. In such cases, the existing method remains in use. However, the role of R&D is continuous improvement iteratively exploring alternative columns (e.g., charged surface hybrid, HILIC), gradient profiles, or detection strategies until a superior modern method is realized.
4. Holistic Justification: The Tangible Return on Modernization
The benefits of this strategic modernization extend far beyond the chromatography data system, impacting the entire organization.
Aspect | Traditional, Fragmented Approach | Modernized, Consolidated Approach | Strategic Impact |
Operational Cost | High solvent and column consumption; frequent repairs due to pressure-induced stress; high waste disposal cost. | 60–80% lower solvent and column usage; reduced instrument downtime; significant waste reduction. | Direct, recurring cost savings; supports environmental sustainability goals. |
Time & Efficiency | Separate, long analytical methods (e.g., 80+ minutes per sample for assay and related substances). | Single, short analytical method (e.g., ~10 minutes per sample); laboratory throughput increases by 5–10×. | Accelerated batch release (mitigates supply-chain risk); elimination of QC bottlenecks; frees analyst time for value-added activities. |
Data Quality & Scientific Rigor | Potential for incomplete impurity profiling; reliance on subjective, classical interpretation. | Higher sensitivity and resolution; objective, automated analysis; robust mass balance; MS-confirmed identification. | Enhanced scientific rigor and product understanding; stronger foundation for regulatory filings and lifecycle management. |
Compliance & Data Integrity | High risk of deviations due to method-induced failures; manual data recording prone to error. | Robust methods reduce failures; electronic data from UPLC and automated titrators ensure full, audit-trailed ALCOA+ compliance. | Lower regulatory risk; fewer investigations; stronger audit defense; transforms data integrity from a challenge into a built-in feature. |
R&D & Lifecycle Role | Method development often siloed and focused only on initial registration requirements. | Strategic method development focused on lifecycle robustness, efficiency, and control from inception. | Transforms QC from a cost center into a strategic asset; embeds continuous improvement across the product lifecycle. |
5. Conclusion: A Strategic Imperative for the Modern Pharma Enterprise
The modernization of pharmaceutical analytical methods from consolidated UPLC protocols and LC-MS identification to automated potentiometry is an urgent strategic imperative, not a discretionary technical upgrade. It represents a fundamental shift from reactive, compliance focused firefighting to proactive, science-driven quality assurance.
This transition is fully supported and encouraged by the modern ICH quality guideline ecosystem (Q2, Q3, Q12, Q14). The one-time investment in re-development, validation, and regulatory submission is decisively outweighed by the perpetual, multiplicative returns: the elimination of costly deviation cycles, dramatic reductions in operational expenses, and the attainment of a higher, more defensible standard of product quality and control.
For well-characterized commercial molecules, this is a proven, globally accepted path to efficiency. For critical molecules, disciplined, R&D-driven improvement remains the path forward. Ultimately, analytical excellence in the 21st century is defined not by the longevity of a legacy method, but by the initiative to modernize leveraging science and technology within regulatory acceptance and approval in the relentless pursuit of greater efficiency, reliability, environmental responsibility, and patient safety.
References
- International Council for Harmonization (ICH). Q2(R2) Guideline: Validation of Analytical Procedures. 2022.
- International Council for Harmonization (ICH). Q3A(R2) Guideline: Impurities in New Drug Substances. 2006.
- International Council for Harmonization (ICH). Q3B(R2) Guideline: Impurities in New Drug Products. 2006.
- International Council for Harmonization (ICH). Q12 Guideline: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management. 2019.
- International Council for Harmonization (ICH). Q14 Guideline: Analytical Procedure Development. 2022.
- United States Pharmacopeia (USP). General Chapter <1225> Validation of Compendial Procedures.
- Food and Drug Administration (FDA). Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics. 2015.
- European Medicines Agency (EMA). Guideline on the use of Near Infrared Spectroscopy by the pharmaceutical industry and the data requirements for new submissions and variations. 2014.
- Snyder, L. R., Kirkland, J. J., & Dolan, J. W. Introduction to Modern Liquid Chromatography. 3rd ed. John Wiley & Sons, 2010.
- Swartz, M. E. Ultra Performance Liquid Chromatography (UPLC): An Introduction. Separation Science Reimagined, 2005.
- Borman, P., et al. "The Application of Quality by Design to Analytical Methods." Pharmaceutical Technology, 31(10), 2007.
- Vishwanathan, K., et al. "Demonstration of Equivalency of a UPLC Method to an HPLC Method for the Determination of Assay and Related Substances in Pharmaceutical Drug Products." Journal of Pharmaceutical and Biomedical Analysis, 51(3), 2010.
- Van Deemter, J. J., Zuiderweg, F. J., & Klinkenberg, A. "Longitudinal diffusion and resistance to mass transfer as causes of nonideality in chromatography." Chemical Engineering Science, 5(6), 1956.
- Anastas, P. T., & Warner, J. C. Green Chemistry: Theory and Practice. Oxford University Press, 1998. (For environmental safety principles).




